PSI/TM-Coffee Tutorial A quick start guide to aligning proteins using homology extension against reduced databases
Description
The web server aligns non-transmembrane and transmembrane proteins (TMPs) [1] by combining homology extension into the T-Coffee consistency approach, termed Position Specific Iterative T-Coffee (PSI-Coffee) [2], and also perform transmembrane topology prediction for TMPs.
TM-Coffee is an alignment procedure that uses reduced reference databases for homology extension to obtain similar results at a significantly reduced computational cost over full protein databases [3]. Our benchmarking on BAliBASE2-ref7 alpha-helical TMPs shows a significant improvement over the most accurate methods [3]. The homology extension and transmembrane topology prediction is implemented in the web server, as well as the T-Coffee package [4].
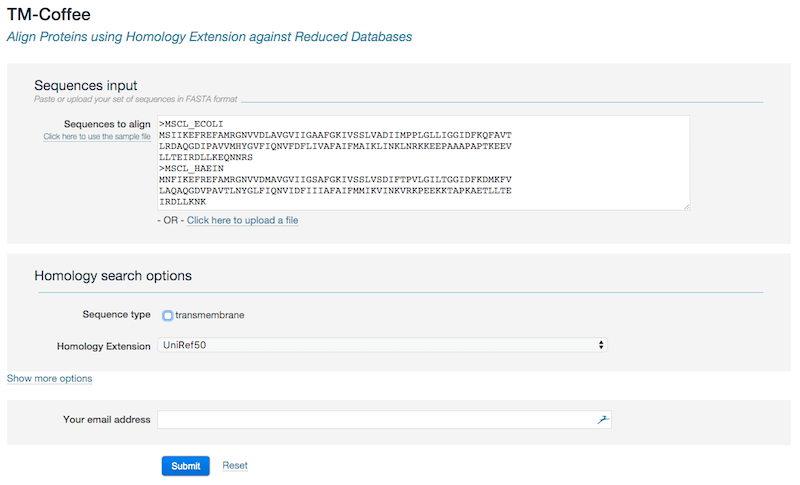
Data input
- Sequences input Enter the sequences in the input field or alternatively provide them using the Upload link. The sequence size limit is 1000 sequences and the sequence length limit is 5000.
- Sequences must be in FASTA format
-
Homology search options Use these options to control the homology search procedure.
- Sequence type: non-transmembrane by default. When checking transmembrane, the source databases will be further reduced by only keeping entried containing "keyword:transmembrane". These TMP specific databases are typically 20% of the size of their sources [3]. Besides performing alignment, the prediction of the transmembrane topology will be conducted for each sequence. The output will be tm_html instead of score_html (see more details in the Output interpretation section).
-
Homology extension databases: you can specify the database which homology extension will be carried out against:
- UniRef50, very fast, rough (default) : built by clustering UniRef100 sequences at the 50% sequence identity levels [5].
- UniRef90, fast, approximate : built by clustering UniRef100 sequences at the 90% sequence identity levels [5].
- UniRef100, slow, accurate : combines identical sequences and sub-fragments from any source organism into a single UniRef entry (i.e., cluster) [5].
- UniProt, slow, accurate
- NCBI NR, slow, accurate
- You may access the advanced settings by clicking the link Show more options. Please refer to the Output options section for detailed information.
- Provide your email address if you want to be notified when the job complete.
- Press the button submit to submit your request.
- Alignment format
- clustalw_aln, pir_aln, pir_seq, gcg, fasta_aln, msf_aln, phylip: several standard alignment formats are supported.
- score_ascii: the plain text format indicating the ResidueTCS, ColumnTCS, SequenceTCS and AlignmentTCS scores, respectively [6].
- Graphic format
- score_html: the html format of score_ascii
- score_pdf: will transfer score_html into pdf format
- Case upper/lower/keep will output the alignment in upper/lower/original case, respectively.
- Residue number on/off will output the alignment with/without the residue numbers beside the alignment, respectively.
- outorder input/aligned will order the sequences of the alignment according to input/align procedure, respectively.
- Alignment length control the length of the alignment per row (default 80).
Please note that the computation can take some minutes to be fulfilled. You don't need to keep your browser open to wait for the result. You can close it or navigate away to another page. Use the History link, in top-bar menu to access the computation result at any moment. You can also retrieve your results using the permanent URL http://tcoffee.crg.cat/apps/tcoffee/result?rid=jobid, where jobid is the ID given at the time of submission.
Output options
Use these options to control the output formats of the alignment 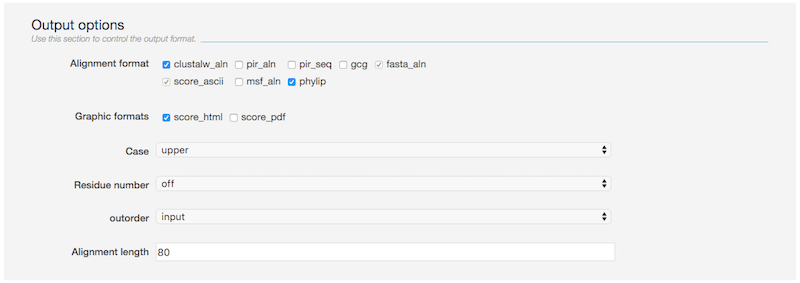
Output interpretation
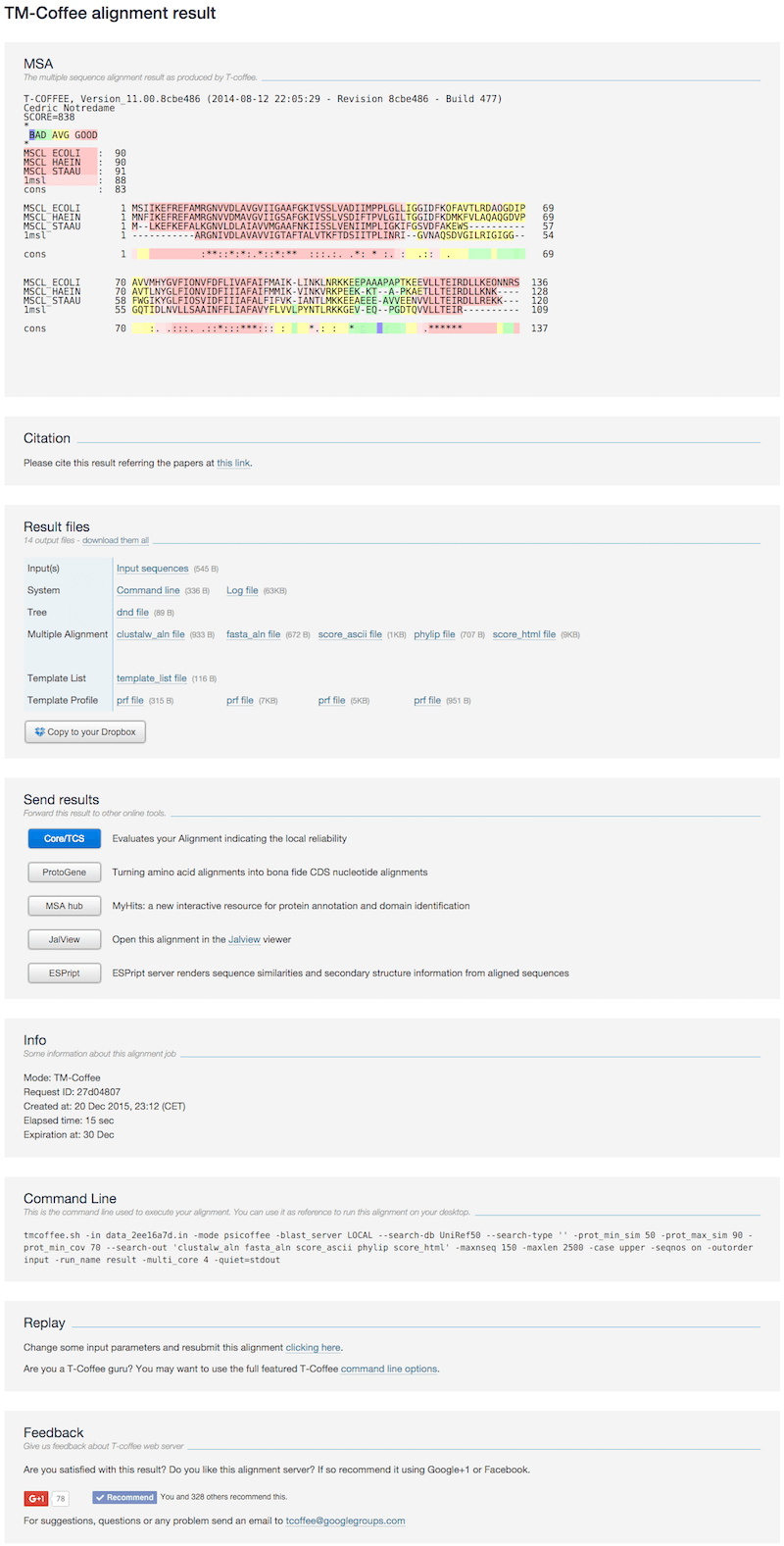
Non-transmembrane proteins
The result page contains the following sections:- MSA The MSA is colored according to the T-Coffee TCS scheme. Dark pink bits are very reliable, while blue and green bits are unreliable based on the T-Coffee library. The color scheme has been redesigned to be easily visualized by color blind people.
- Citation The related article when citing the TM-Coffee.
- Result files
- Tree the guide tree used during the alignment procedure.
- Multiple Alignment subsection provides the MSAs in CLUSTAL, FASTA and Phylip formats. score_html is the html format of the above MSA section and its plain text format, score_ascii.
- Template List lists the homology profiles for sequences
- Template Profile the homology profile of each sequence from BLAST search procedure
- Send results Forward this result to other online tools
- Info Information related to the submitted job (running time, date, etc).
- Replay This link allows the user to re-run the job while modifying input options or data.
- Feedback To share your alignment experience on social networks.
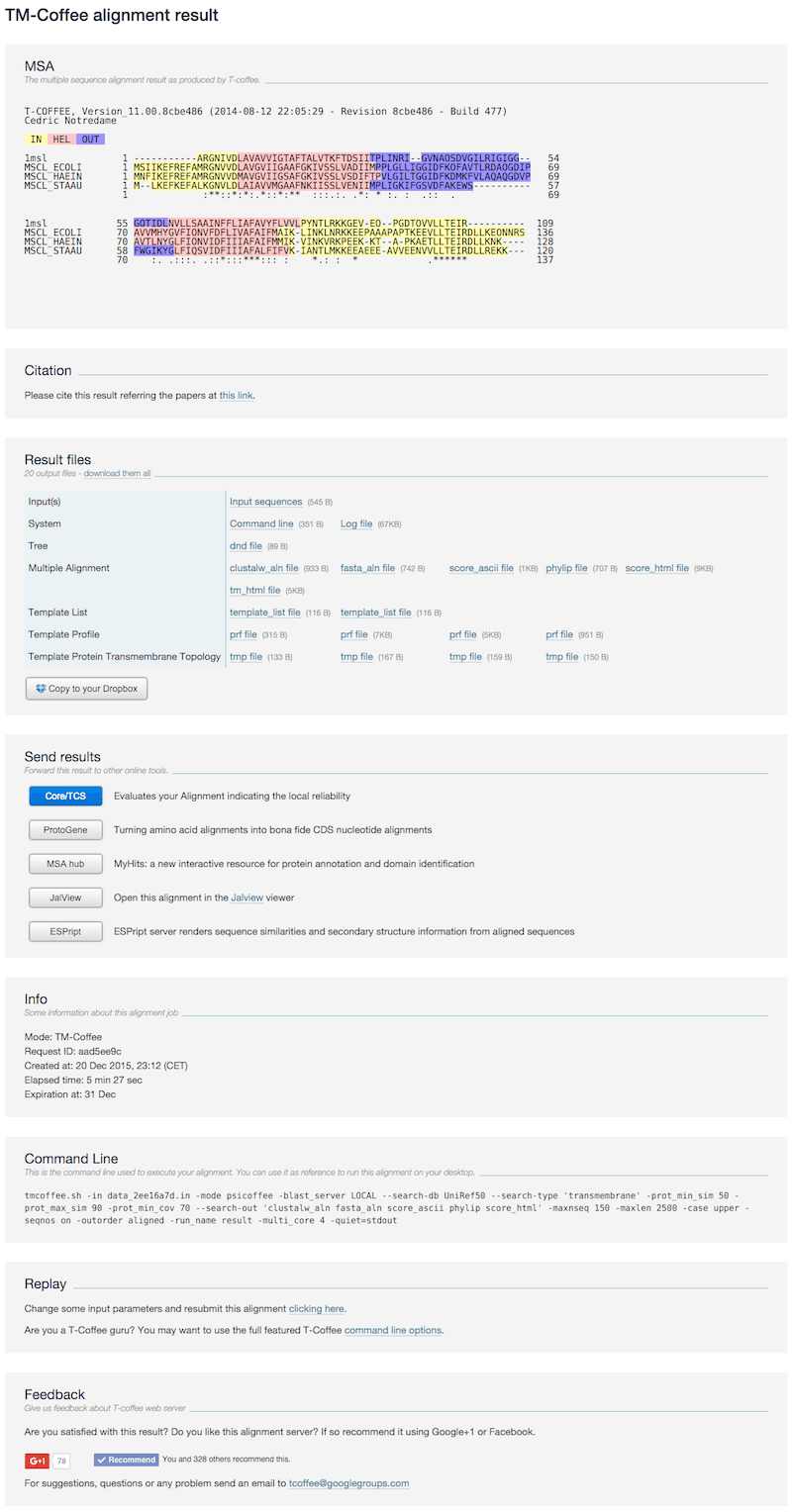
Transmembrane proteins
The result page contains the following sections:- MSA The MSA is colored according to transmembrane topology prediction by HMMTOP [7], where yellow: in loop, red: TM helix and blue: out loop
- Citation The related article when citing the TM-Coffee.
- Result files
- Tree the guide tree used during the alignment procedure
- Multiple Alignment subsection provides the MSAs in CLUSTAL, FASTA and Phylip formats. score_html the html format of the T-Coffee TCS score and its plain text format, score_ascii. tm_html the html format of the above MSA section indicates the transmembrane topology prediction.
- Template List lists the homology profiles and topology predictions for sequences
- Template Profile the homology profile of each sequence from the BLAST search procedure
- Template Protein Transmembrane Topology the transmembrane topology prediction of each sequence, where I: in loop, H: TM helix and O: out loop
- Send results Forward this result to other online tools
- Info Information related to the submitted job (running time, date, etc).
- Replay This link allows the user to re-run the job while modifying input options or data.
- Feedback To share your alignment experience on social networks.
Supplemental Data
- PSI/TM-Coffee: a web server for accurate multiple sequence alignment of (non-)transmembrane proteins by using homology information.
-
Upcoming challenges for multiple sequence alignment methods in the high-throughput era
Kemena C., Notredame C.
Bioinformatics 25, 2455–65 (2009). (PMID: 19648142) -
Accurate multiple sequence alignment of transmembrane proteins with PSI-Coffee
Jia-Ming Chang, Paolo Di Tommaso, Jean-François Taly and Cedric Notredame
BMC Bioinformatics 13, S1 (2012). (PMID: 22536955) - PSI-Coffee stand alone usage
-
UniRef clusters: a comprehensive and scalable alternative for improving sequence similarity searches
Suzek BE, Wang Y, Huang H, McGarvey PB, Wu CH; UniProt Consortium
Bioinformatics (2014). (PMID: 25398609) -
TCS: a new multiple sequence alignment reliability measure to estimate alignment accuracy and improve phylogenetic tree reconstruction
Chang JM, Di Tommaso P, Notredame C.
Molecular Biology and Evolution 31, 1625–37 (2014). (PMID: 24694831) -
The HMMTOP transmembrane topology prediction server
Tusnády GE, Simon I
Bioinformatics 17, 849–50 (2001). (PMID: 11590105)